Play Video

reproduce el video
What is Alagille Syndrome?
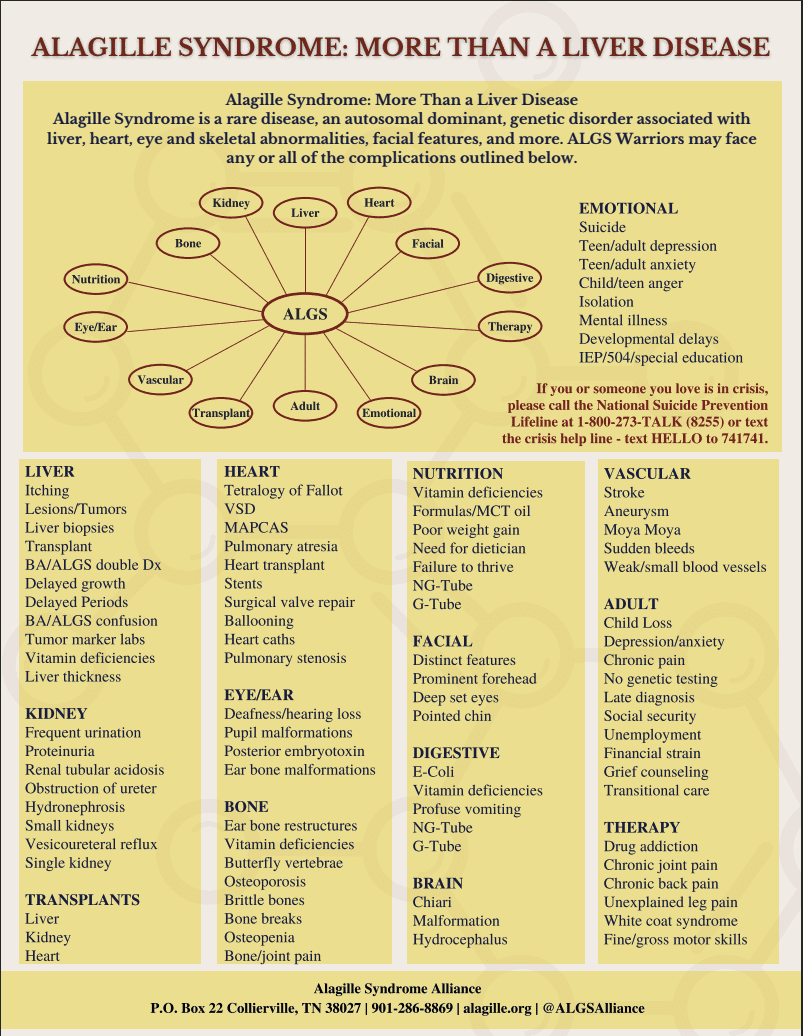
Alagille Syndrome (OMIM #118450) is the most commonly encountered rare cholestatic liver disease seen by pediatric hepatologists. It can affect multiple organ systems of the body including the liver, heart, skeleton, eyes and kidneys. The specific symptoms and severity of Alagille syndrome can vary greatly from one person to another, even within the same family. Some individuals may have mild forms of the disorder while others may have more serious forms.
Most people with Alagille syndrome have mutations in one copy of the JAG1 gene. A small percentage (less than 1 percent) of patients have mutations of the NOTCH2 gene. These mutations are inherited as autosomal dominant traits, however in about half of cases the mutation occurs as a new change (“de novo”) in the individual and was not inherited from a parent. The current estimated incidence of ALGS is between 1:30,000 and 1:70,000 with no difference in gender.
The video to right, in English and Spanish, is a wonderful animated description of Alagille Syndrome. Sponsored by Travere Therapeutics for the ALGS community, it was developed by National Organization for Rare Disorders (NORD).
Below, you will find frequently asked questions about Alagille Syndrome which will continue to be expanded through 2024.
Play Video
reproduce el video
Adults with ALGS
Most people with Alagille syndrome have mutations in one copy of the JAG1 gene. A small percentage (less than 1 percent) of patients have mutations of the NOTCH2 gene. These mutations are inherited as autosomal dominant traits, however in about half of cases the mutation occurs as a new change (“de novo”) in the individual and was not inherited from a parent. The current estimated incidence of ALGS is between 1:30,000 and 1:70,000 with no difference in gender.
Click on any of the images on the right hand side of this page to view a very informative video about Alagille Syndrome.
Recommended Reading



Frequently Asked Questions
How is Alagille syndrome diagnosed?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
How is Alagille syndrome diagnosed?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Is there a cure for Alagille syndrome?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Is Alagille syndrome hereditary?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
How is Alagille syndrome diagnosed?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
What are the symptoms / characteristics of Alagille Syndrome?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Are there treatments for Alagille syndrome?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
Is Alagille syndrome life limiting or life threatening?
Your content goes here. Edit or remove this text inline or in the module Content settings. You can also style every aspect of this content in the module Design settings and even apply custom CSS to this text in the module Advanced settings.
ALGS Specialist
| wdt_ID | Country | IMage | Image | Program Name (Pediatrics) | Location Details | Important Links |
|---|---|---|---|---|---|---|
| 1 | USA |
 |
 |
Program Name (Pediatrics) Alagille Syndrome Program 650-399-3765 Alagille@stanfordchildrens.org |
Location Details Lucile Packard Children’s Hospital Stanford 725 Welch Rd State: California Palo Alto, CA 94304 |
Important Links Alagille Syndrome Program |
| 2 | USA |
 |
Program Name (Pediatrics) Alagille Syndrome Program 650-399-3765 Alagille@stanfordchildrens.org |
Location Details Lucile Packard Children’s Hospital Stanford 725 Welch Rd Palo Alto, CA 94304 |
Important Links Liver Clinic |
|
| 3 | USA |
 |
Program Name (Pediatrics) Gastroenterology, Hepatology & Nutrition 323-361-2181 |
Location Details Dividion of Gastroenterology, Hepatology & Nutrition 4650 Sunset Blvd. State: Los Angeles CA 90027 |
Important Links Gastroenterology, Hepatology Nutrition |
|
| 4 | USA |
 |
Program Name (Pediatrics) The UCLA Health Pediatric Liver Transplant Program 310-825-8138 |
Location Details UCLA Mattel Children’s Hospital 757 Westwood Plaza State: Los Angeles CA 90095 |
Important Links Pediatric Liver Transplant Program Liver Transplant Expert Team |
|
| 5 | USA |
 |
Program Name (Pediatrics) Pediatric Liver Center 877-822-4453 |
Location Details UCSF Benioff Children’s Hospitals 744 52nd St. State: Oakland CA 94609 |
Important Links Pediatric Liver Center Pediatric Liver Transplant Program |

Disease Education

-
 Alagille Syndrome Alliance
Alagille Syndrome Alliance
ALGSAcademy

-
National Organization for Rare Disorders

NORD Alagille Syndrome Explanation Video
ALGS education is crucial for understanding the unique complexity of Alagille Syndrome. The reasons behind its varying severity remain unclear, making accessible resources essential. Explore our updated library covering diagnosis, ALGS in the classroom, genetics, and more.

-
Karger
Karger Learning Module for Physicians and Patients

-
HCP live
Peer Exchange: Management of ALGS

-
Rare Disease 360

Rare Disease 360 – Alagille Syndrome Channel

Resource Library

Patient and Caregiver Manuals and Guides

One-Page Educational Resources

Translated Resources

ALGSA Patient and Family Program Guide

New Diagnosis Guide

ALGS Care Manual

Succeeding in the Classroom Guide

What is Alagille Syndrome

The Genetics of ALGS

Genetics of ALGS: FAQ